Help
Navigation
Navigating by Gene Letter
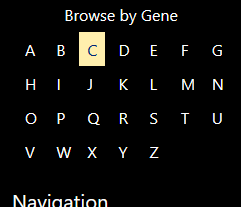
When you click a letter on the table to the left, you will be shown all genes starting with that letter that have variants associated with them.

The Gene Page
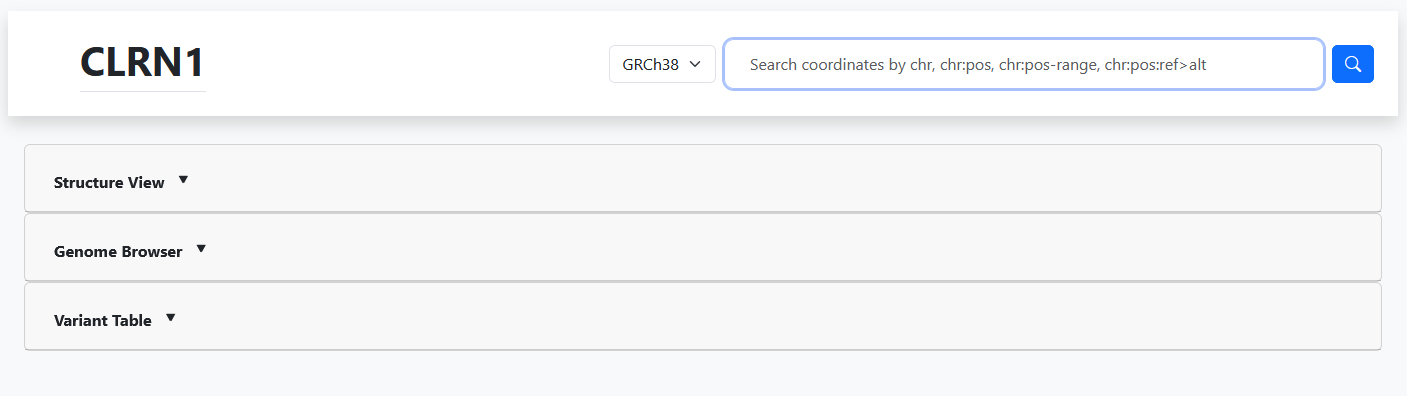 The Gene Page offers gene-level information for review on the webpage as well as filterable and downloadable variant lists.
On this webpage, you can view all variants associated with that gene, a CADD Phred score boxplot of pathogenicity classifications for that gene, the gene's variants in an interactive genomic browser, and view the gene's 3D protein structure.
For detailed information regarding the functionality of these interactive viewers, please see their individual sections of this help page:
The Gene Page offers gene-level information for review on the webpage as well as filterable and downloadable variant lists.
On this webpage, you can view all variants associated with that gene, a CADD Phred score boxplot of pathogenicity classifications for that gene, the gene's variants in an interactive genomic browser, and view the gene's 3D protein structure.
For detailed information regarding the functionality of these interactive viewers, please see their individual sections of this help page:
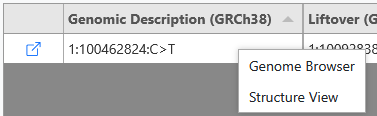
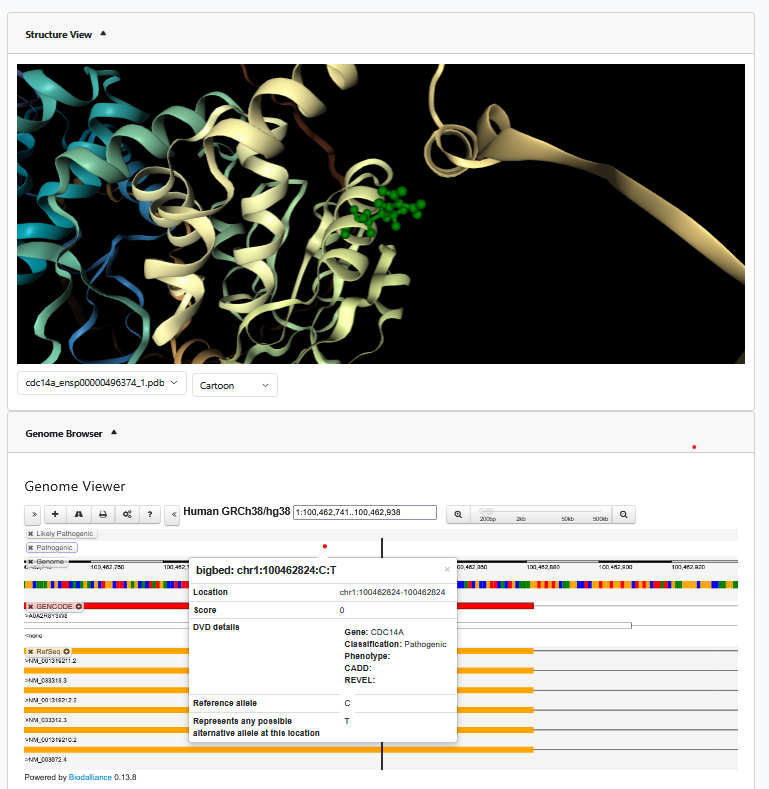
A variant of interest can be viewed on the genome browser or the protein structure veiw by right clicking on the variant row in the table.
The coordinates are automatically changed to the variant position.
Navigating by the Variant Search Bar
The search bar featured at the top of most MORL pages enables you to search for a single specific variant, all variants at a position, all variants within a range of positions along the same chromosome, or any combination of those three options. The search bar hooks into the API so that a search of 13:20763071:T>C is equivalent to https://deafnessvariationdatabase.org/api?version=9.2&type=hg19coord&method=IO&format=web&terms=13:20763071:T>C.
For more information about the supporting API call, please review the API Documentation page.
The expected return of the search bar is a loading of a new page where,
- all of the search terms are displayed under the heading "Terms searched:"
- all of the individual searched terms are displayed as faux-gene headings with API links to their variant list results, similar to the gene page variant lists.
- display a loading wait bar with status messages for data retrieval.
- generate a table to view variant data grouped by gene.
- Show a grey message of "Loaded" or "No Data" to the right of the clicked heading.
Single Search Term Examples:
- Specific variant search -
13:20763071:T>C - Variants at a position -
13:20763044 - Variants within a range -
13:20763045-20763071 - Not found -
X:79476000
The search bar accepts independent searches separated by commas. Spaces are ignored.
Multiple Search Term Examples:
- Specific variant and position search -
13:20763071:T>C, 13:20763044 - Range and position search -
13:20763044, 13:20763045-20763071
Getting Support
Please get in touch if you experience any issues with using this site.